
Ultrahigh specificity in a network of computationally designed protein-interaction pairs
Ravit Netzer, Dina Listov, Rosalie Lipsh and Sarel J. Fleishman
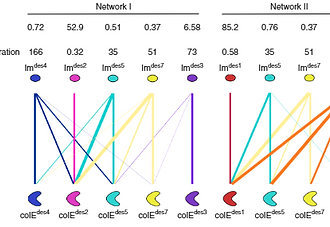
Protein networks in all organisms comprise homologous interacting pairs. In these networks,some proteins are specific, interacting with one or a few binding partners, whereas others are multispecific and bind a range of targets. We describe an algorithm that starts from an interacting pair and designs dozens of new pairs with diverse backbone conformations at the binding site as well as new binding orientations and sequences. Applied to a high-affinity bacterial pair, the algorithm results in 18 new ones, with cognate affinities from pico- to micromolar. Three pairs exhibit 3-5 orders of magnitude switch in specificity relative to the wild type, whereas others are multispecific, collectively forming a protein-interaction net-work. Crystallographic analysis confirms design accuracy, including in new backbones and polar interactions. Preorganized polar interaction networks are responsible for high specificity, thus defining design principles that can be applied to program synthetic cellular inter-action networks of desired affinity and specificity

Automated Design of Efficient and Functionally Diverse Enzyme Repertoires
Olga Khersonsky, Rosalie Lipsh, Ziv Avizemer and Sarel J. Fleishman
Substantial improvements in enzyme activity demand multiple mutations at spatially proximal positions in the active site. Such mutations, however, often exhibit unpredictable epistatic (non-additive) effects on activity. Here we describe FuncLib, an automated method for designing multipoint mutations at enzyme active sites using phylogenetic analysis and Rosetta design calculations. We applied FuncLib to two unrelated enzymes, a phosphotriesterase and an acetyl-CoA synthetase. All designs were active, and most showed activity profiles that significantly differed from the wild-type and from one another. Several dozen designs with only 3–6 active-site mutations exhibited 10- to 4,000-fold higher efficiencies with a range of alternative substrates, including hydrolysis of the toxic organophosphate nerve agents soman and cyclosarin and synthesis of butyryl-CoA. FuncLib is implemented as a web server (http://FuncLib.weizmann.ac.il); it circumvents iterative, high-throughput experimental screens and opens the way to designing highly efficient and diverse catalytic repertoires.
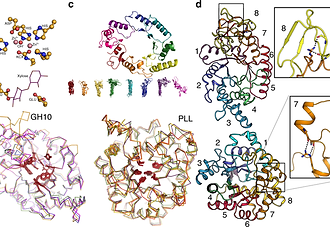
Highly active enzymes by automated combinatorial backbone assembly and sequence design
Gideon Lapidoth, Olga Khersonsky, Rosalie Lipsh, and Sarel Fleishman et al, Nature communications,2018
Automated design of enzymes with wild-type-like catalytic properties has been a longstanding but elusive goal. Here, we present a general, automated method for enzyme design through combinatorial backbone assembly. Starting from a set of homologous yet structurally diverse enzyme structures, the method assembles new backbone combinations and uses Rosetta to optimize the amino acid sequence, while conserving key catalytic residues. We apply this method to two unrelated enzyme families with TIM-barrel folds, glycoside hydrolase 10 (GH10) xylanases and phosphotriesterase-like lactonases (PLLs), designing 43 and 34 proteins, respectively. Twenty-one GH10 and seven PLL designs are active, including designs derived from templates with <25% sequence identity. Moreover, four designs are as active as natural enzymes in these families. Atomic accuracy in a high-activity GH10 design is further confirmed by crystallographic analysis. Thus, combinatorial-backbone assembly and design may be used to generate stable, active, and structurally diverse enzymes with altered selectivity or activity.
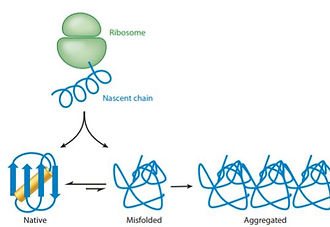
Principles of Protein Stability and Their Application in Computational Design
Adi Goldenzweig and Sarel Fleishman,Annual Review of Biochemistry,2018
Proteins are increasingly used in basic and applied biomedical research.Many proteins, however, are only marginally stable and can be expressed in limited amounts, thus hampering research and applications. Research has revealed the thermodynamic, cellular, and evolutionary principles and mechanisms that underlie marginal stability. With this growing understanding, computational stability design methods have advanced over the past two decades starting from methods that selectively addressed only some aspects of marginal stability. Current methods are more general and, by combining phylogenetic analysis with atomistic design, have shown drastic improvements in solubility, thermal stability, and aggregation resistance while maintaining the protein’s primary molecular activity. Stability design is opening the way to rational engineering of improved enzymes, therapeutics, and vaccines and to the application of protein design methodology to large proteins and molecular activities that have proven challenging in the past.
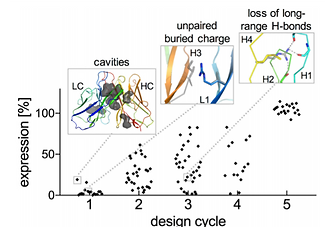
Principles for computational design of binding antibodies
Dror Baran, M. Gabriele Pszolla, Sarel Fleishman, et al, PNAS, 2017
Natural proteins must both fold into a stable conformation and exert their molecular function. To date, computational design has successfully produced stable and atomically accurate proteins by using so-called “ideal” folds rich in regular secondary structures and almost devoid of loops and destabilizing elements, such as cavities. Molecular function, such as binding and catalysis, however, often demands nonideal features, including large and irregular loops and buried polar interaction networks, which have remained challenging for fold design. Through five design/experiment cycles, we learned principles for designing stable and functional antibody variable fragments (Fvs). Specifically, we (i) used sequence-design constraints derived from antibody multiple-sequence alignments, and (ii) during backbone design, maintained stabilizing interactions observed in natural antibodies between the framework and loops of complementarity-determining regions (CDRs) 1 and 2. Designed Fvs bound their ligands with midnanomolar affinities and were as stable as natural antibodies, despite having >30 mutations from mammalian antibody germlines. Furthermore, crystallographic analysis demonstrated atomic accuracy throughout the framework and in four of six CDRs in one design and atomic accuracy in the entire Fv in another. The principles we learned are general, and can be implemented to design other nonideal folds, generating stable, specific, and precise antibodies and enzymes
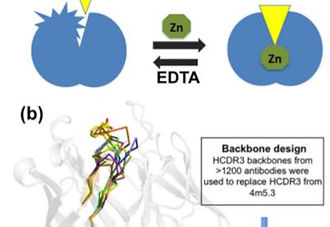
Incorporating an allosteric regulatory site in an antibody through backbone design
Olga Khersonsky and Sarel Fleishman, Protein Science, 2017
Allosteric regulation underlies living cells’ ability to sense changes in nutrient and
signaling-molecule concentrations, but the ability to computationally design allosteric regulation
into non-allosteric proteins has been elusive. Allosteric-site design is complicated by the require-
ment to encode the relative stabilities of active and inactive conformations of the same protein in
the presence and absence of both ligand and effector. To address this challenge, we used Rosetta
to design the backbone of the flexible heavy-chain complementarity-determining region 3 (HCDR3),
and used geometric matching and sequence optimization to place a Zn 21 -coordination site in a
fluorescein-binding antibody. We predicted that due to HCDR3’s flexibility, the fluorescein-binding
pocket would configure properly only upon Zn 21 application. We found that regulation by Zn 21 was
reversible and sensitive to the divalent ion’s identity, and came at the cost of reduced antibody sta-
bility and fluorescein-binding affinity. Fluorescein bound at an order of magnitude higher affinity in
the presence of Zn 21 than in its absence, and the increase in fluorescein affinity was due almost
entirely to faster fluorescein on-rate, suggesting that Zn 21 preorganized the antibody for fluoresce-
in binding. Mutation analysis demonstrated the extreme sensitivity of Zn 21 regulation on the atomic
details in and around the metal-coordination site. The designed antibody could serve to study how
allosteric regulation evolved from non-allosteric binding proteins, and suggests a way to designing
molecular sensors for environmental and biomedical targets.
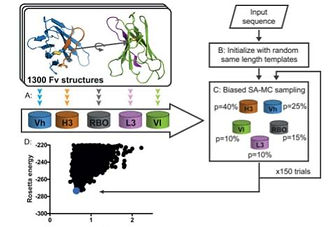
High-accuracy modeling of antibody structures by a search for minimum-energy recombination of backbone fragments
Christoffer H. Norn, Gideon Lapidoth, and Sarel J. Fleishman, Proteins, 2017
Current methods for antibody structure prediction rely on sequence homology to known structures. Although this strategy often yields accurate predictions, models can be stereo-chemically strained. Here, we present a fully automated algorithm, called AbPredict, that disregards sequence homology, and instead uses a Monte Carlo search for low-energy conformations built from backbone segments and rigid-body orientations that appear in antibody molecular structures. We find cases where AbPredict selects accurate loop templates with sequence identity as low as 10%, whereas the template of highest sequence identity diverges substantially from the query’s conformation. Accordingly, in several cases reported in the recent Antibody Modeling Assessment benchmark, AbPredict models were more accurate than those from any participant, and the models’ stereo-chemical quality was consistently high. Furthermore, in two blind cases provided to us by crystallographers prior to structure determination, the method achieved <1.5 A˚ overall backbone accuracy. Accurate modeling of unstrained antibody structures will enable design and engineering of improved binders for biomedical research directly from sequence.

One-step design of a stable variant of the malaria invasion protein RH5 for use as a vaccine immunogen
Ivan Campeottoa, Adi Goldenzweig, Matthew K. Higgins, Sarel Fleishman, et al PNAS, 2017
Many promising vaccine candidates from pathogenic viruses, bacteria, and parasites are unstable and cannot be produced cheaply for clinical use. For instance, Plasmodium falciparum reticulocyte-binding protein homolog 5 (PfRH5) is essential for erythrocyte invasion, is highly conserved among field isolates, and elicits antibodies that neutralize in vitro and protect in an animal model, making it a leading malaria vaccine candidate. However, functional RH5 is only expressible in eukaryotic systems and exhibits moderate temperature tolerance, limiting its usefulness in hot and low-income countries where malaria prevails. Current approaches to immunogen stabilization involve iterative application of rational or semirational design, random mutagenesis, and biochemical characterization. Typically, each round of optimization yields minor improvement in stability, and multiple rounds are required. In contrast, we developed a onestep design strategy using phylogenetic analysis and Rosetta atomistic calculations to design PfRH5 variants with improved packing and surface polarity. To demonstrate the robustness of this approach, we tested three PfRH5 designs, all of which showed improved stability relative to wild type. The best, bearing 18 mutations relative to PfRH5, expressed in a folded form in bacteria at >1 mg of protein per L of culture, and had 10–15 °C higher thermal tolerance than wild type, while also retaining ligand binding and immunogenic properties indistinguishable from wild type, proving its value as an immunogen for a future generation of vaccines against the malaria blood stage. We envision that this efficient computational stability design methodology will also be used to enhance the biophysical properties of other recalcitrant vaccine candidates from emerging pathogens.
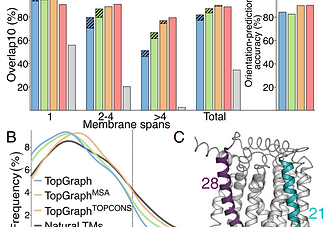
Interplay between hydrophobicity and the positiveinside rule in determining membrane-protein topology
Assaf Elazar, Jonathan Weinstein, Jaime Prilusky, and Sarel Fleishman, PNAS, 2016
The energetics of membrane-protein interactions determine protein topology and structure: hydrophobicity drives the insertion of helical segments into the membrane, and positive charges orient the protein with respect to the membrane plane according to the positive-inside rule. Until recently, however, quantifying these contributions met with difficulty, precluding systematic analysis of the energetic basis for membrane-protein topology. We recently developed the dsTβL method, which uses deep sequencing and in vitro selection of segments inserted into the bacterial plasma membrane to infer insertion-energy profiles for each amino acid residue across the membrane, and quantified the insertion contribution from hydrophobicity and the positive-inside rule. Here, we present a topology-prediction algorithm called TopGraph, which is based on a sequence search for minimum dsTβL insertion energy. Whereas the average insertion energy assigned by previous experimental scales was positive (unfavorable), the average assigned by TopGraph in a nonredundant set is −6.9 kcal/mol. By quantifying contributions from both hydrophobicity and the positive-inside rule we further find that in about half of large membrane proteins polar segments are inserted into the membrane to position more positive charges in the cytoplasm, suggesting an interplay between these two energy contributions. Because membrane-embedded polar residues are crucial for substrate binding and conformational change, the results implicate the positive-inside rule in determining the architectures of membrane-protein functional sites. This insight may aid structure prediction, engineering, and design of membrane proteins. TopGraph is available online (topgraph.weizmann.ac.il).

Automated Structure and Sequence Based Design of Proteins for High Bacterial Expression and Stability
Adi Goldenzweig and Sarel J. Fleishman, et al. Mol. Cell, 2016
Upon heterologous overexpression, many proteins misfold or aggregate, thus resulting in low functional yields. Human acetylcholinesterase (hAChE), an enzyme mediating synaptic transmission, is a typical case of a human protein that necessitates mammalian systems to obtain functional expression. We developed a computational strategy and designed an AChE variant bearing 51 mutations that improved core packing, surface polarity, and backbone rigidity. This variant expressed at ∼2,000-fold higher levels in E. coli compared to wild-type hAChE and exhibited 20°C higher thermostability with no change in enzymatic properties or in the active-site configuration as determined by crystallography. To demonstrate broad utility, we similarly designed four other human and bacterial proteins. Testing at most three designs per protein, we obtained enhanced stability and/or higher yields of soluble and active protein in E. coli.


Why reinvent the wheel? Building new proteins based on ready-made parts
Olga Khersonsky and Sarel J. Fleishman, Protein Science, 2016
We protein engineers are ambivalent about evolution: on the one hand, evolution inspires us with myriad examples of biomolecular binders, sensors, and catalysts; on the other hand, these examples are seldom well-adapted to the engineering tasks we have in mind. Protein engineers have therefore modified natural proteins by point substitutions and fragment exchanges in an effort to generate new functions. A counterpoint to such design efforts, which is being pursued now with greater success, is to completely eschew the starting materials provided by nature and to design new protein functions from scratch by using de novo molecular modeling and design. While important progress has been made in both directions, some areas of protein design are still beyond
reach. To this end, we advocate a synthesis of these two strategies: by using design calculations to both recombine and optimize fragments from natural proteins, we can build stable and as of yet unsampled structures, thereby granting access to an expanded repertoire of conformations and desired functions. We propose that future methods that combine phylogenetic analysis, structure and sequence bioinformatics, and atomistic modeling may well succeed where any one of these approaches has failed on its own.

Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane
Elazar, A. et al. eLIFE ,2016
Insertion of helix-forming segments into the membrane and their association determines the structure, function, and expression levels of all plasma membrane proteins. However, systematic and reliable quantification of membrane-protein energetics has been challenging. We developed a deep mutational scanning method to monitor the effects of hundreds of point mutations on helix insertion and self-association within the bacterial inner membrane. The assay quantifies insertion energetics for all natural amino acids at 27 positions across the membrane, revealing that the hydrophobicity of biological membranes is significantly higher than appreciated. We further quantitate the contributions to membrane-protein insertion from positively charged residues at the cytoplasm-membrane interface and reveal large and unanticipated differences among these residues. Finally, we derive comprehensive mutational landscapes in the membrane domains of Glycophorin A and the ErbB2 oncogene, and find that insertion and self-association are strongly coupled in receptor homodimers.

AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences
Lapidoth, G. D. et al. Proteins, 2015
Computational design of protein function has made substantial progress, generating new enzymes, binders, inhibitors, and nanomaterials not previously seen in nature. However, the ability to design new protein backbones for function—essential to exert control over all polypeptide degrees of freedom—remains a critical challenge. Most previous attempts to design new backbones computed the mainchain from scratch. Here, instead, we describe a combinatorial backbone and sequence optimization algorithm called AbDesign, which leverages the large number of sequences and experimentally determined molecular structures of antibodies to construct new antibody models, dock them against target surfaces and optimize their sequence and backbone conformation for high stability and binding affinity. We used the algorithm to produce antibody designs that target the same molecular surfaces as nine natural, high-affinity antibodies; in five cases interface sequence
identity is above 30%, and in four of those the backbone conformation at the core of the antibody binding surface is within 1 A ̊ root-mean square deviation from the natural antibodies. Designs recapitulate polar interaction networks observed in natural complexes, and amino acid sidechain rigidity at the designed binding surface, which is likely important for affinity and specificity, is high compared to previous design studies. In designed anti-lysozyme antibodies, complementarity-determining regions (CDRs) at the periphery of the interface, such as L1 and H2, show greater backbone conformation diversity than the CDRs at the core of the interface, and increase the binding surface area compared to the natural antibody, potentially enhancing affinity and specificity.
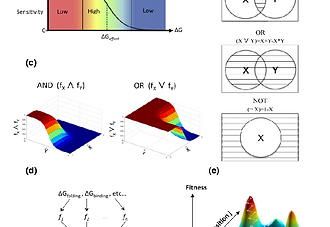
A “Fuzzy”-Logic Language for Encoding Multiple Physical Traits in Biomolecules
Warszawski, S., Netzer, R., Tawfik, D. S. & Fleishman, S. J., Molecular Biology, 2014
To carry out their activities, biological macromolecules balance different physical traits, such as stability, interaction affinity, and selectivity. How such often opposing traits are encoded in a macromolecular system is critical to our understanding of evolutionary processes and ability to design new molecules with desired functions. We present a framework for constraining design simulations to balance different physical characteristics. Each trait is represented by the equilibrium fractional occupancy of the desired state relative to its alternatives, ranging from none to full occupancy, and the different traits are combined using Boolean operators to effect a “fuzzy”-logic language for encoding any combination of traits. In another paper, we presented a new combinatorial backbone design algorithm AbDesign where the fuzzy-logic framework was used to optimize protein backbones and sequences for both stability and binding affinity in antibody-design simulation. We now extend this framework and find that fuzzy-logic design simulations reproduce sequence and structure design principles seen in nature to underlie exquisite specificity on the one hand and multispecificity on the other hand. The fuzzy-logic language is broadly applicable and could help define the space of tolerated and beneficial mutations in natural biomolecular systems and design artificial molecules that encode complex characteristics.
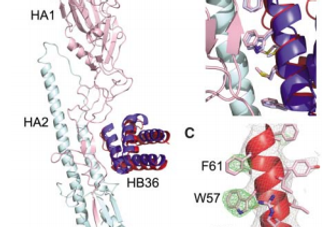
Computational Design of Proteins Targeting the Conserved Stem Region of Influenza Hemagglutinin
Sarel J. Fleishman,David Baker, Science, 2011
We describe a general computational method for designing proteins that bind a surface patch of interest on a target macromolecule. Favorable interactions between disembodied amino acid residues and the target surface are identified and used to anchor de novo designed interfaces. The method was used to design proteins that bind a conserved surface patch on the stem of the influenza hemagglutinin (HA) from the 1918 H1N1 pandemic virus. After affinity maturation, two of the designed proteins, HB36 and HB80, bind H1 and H5 HAs with low nanomolar affinity. Further, HB80 inhibits the HA fusogenic conformational changes induced at low pH. The crystal structure of HB36 in complex with 1918/H1 HA revealed that the actual binding interface is nearly identical to that in the computational design model. Such designed binding proteins may be useful for both diagnostics and therapeutics.

Role of the Biomolecular Energy Gap in Protein Design, Structure, and Evolution
S. J. Fleishman and D. Baker, Cell, 2012
The folding of natural biopolymers into unique three-dimensional structures that determine their function is remarkable considering the vast number of alternative states and requires a large gap in the energy of the functional state compared to the many alternatives. This Perspective explores the implications of this energy gap for computing the structures of naturally occurring biopolymers, designing proteins with new structures and functions, and optimally integrating experiment and computation in these endeavors. Possible parallels between the generation of functional molecules in computational design and natural evolution are highlighted.

Hotspot-Centric De Novo Design of Protein Binders
Fleishman, S. J. et al. J. Molecular Biology, 2011
Protein–protein interactions play critical roles in biology, and computa- tional design of interactions could be useful in a range of applications. We describe in detail a general approach to de novo design of protein interactions based on computed, energetically optimized interaction hotspots, which was recently used to produce high-affinity binders of influenza hemagglutinin. We present several alternative approaches to identify and build the key hotspot interactions within both core secondary structural elements and variable loop regions and evaluate the method's performance in natural-interface recapitulation. We show that the method generates binding surfaces that are more conformationally restricted than previous design methods, reducing opportunities for off-target interactions.

Computational Design of Novel Protein Binders and Experimental Affinity Maturation
Whitehead, T. a, Baker, D. & Fleishman, S. J. Methods Enzymology, 2013
Computational design of novel protein binders has recently emerged as a useful technique to study biomolecular recognition and generate molecules for use in biotechnol- ogy, research, andbiomedicine.Current limitations in computational design methodology have led to the adoption of high-throughput screening and affinity maturation techniques to diagnose modeling inaccuracies and generate high activity binders. Here, we scrutinize this combination of computational and experimental aspects and propose areas for future methodological improvements.
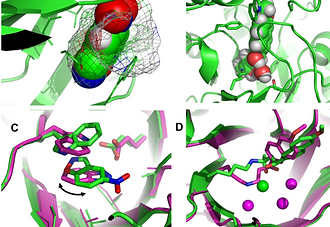
Emerging themes in the computational design of novel enzymes and protein–protein interfaces
Khare, S. D. & Fleishman, S. J. FEBS Letters, 2013
Recent years have seen the first applications of computational protein design to generate novel cat- alysts, binding pairs of proteins, protein inhibitors, and large oligomeric assemblies. At their core these methods rely on a similar hybrid energy function, composed of physics-based and database- derived terms, while different sequence and conformational sampling approaches are used for each design category. Although these are first steps for the computational design of novel function, crystal structures and biochemical characterization already point out where success and failure are likely in the application of protein design. Contrasting failed and successful design attempts has been used to diagnose deficiencies in the approaches and in the underlying hybrid energy function. In this manner, design provides an inherent mechanism by which crucial information is obtained on pressing areas where focused efforts to improve methods are needed. Of the successful designs, many feature pre-organized sites that are poised to perform their intended function, and improvements often result from disfavoring alternative functionally suboptimal states. These rapid develop- ments and fundamental insights obtained thus far promise to make computational design of novel molecular function general, robust, and routine.
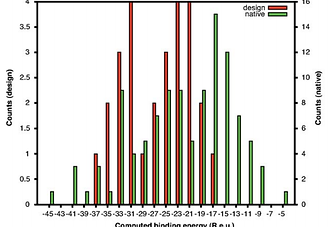
Restricted sidechain plasticity in the structures of native proteins and complexes
Fleishman, S. J., Khare, S. D., Koga, N. & Baker, D. Protein Science, 2013
Protein-design methodology can now generate models of protein structures and interfaces with computed energies in the range of those of naturally occurring structures. Comparison of the properties of native structures and complexes to isoenergetic design models can provide insight into the properties of the former that reflect selection pressure for factors beyond the energy of the native state. We report here that sidechains in native structures and interfaces are significantly more constrained than designed interfaces and structures with equal computed binding energy or stability, which may reflect selection against potentially deleterious non-native interactions.